
Protein aggregation:
Toxicity and function, two sides of the same coin
 Protein misfolding and aggregation are linked to the onset of more than 40 human diseases. Although protein aggregation is potentially harmful for the cell and usually compromises its fitness, the vast majority of proteins contain sequences that predispose them to aggregate. The reason behind this apparent contradiction is that the presence of such sequences provides a number of structural and functional advantages, as long as they are kept under control.
Protein misfolding and aggregation are linked to the onset of more than 40 human diseases. Although protein aggregation is potentially harmful for the cell and usually compromises its fitness, the vast majority of proteins contain sequences that predispose them to aggregate. The reason behind this apparent contradiction is that the presence of such sequences provides a number of structural and functional advantages, as long as they are kept under control.
A growing interest in the study of protein aggregation
The study of protein aggregation has remained for long time as a marginal research topic, restricted to the biotechnological area. Protein aggregates were only of interest as a source of recombinant proteins during the heterologous expression of insoluble proteins and most of the efforts in this field were devoted to find conditions in which they could be refolded to render the maximal yield of native protein, usually via trial and error approaches [1]. The revival of protein aggregation during the last two decades owes to the discovery that this phenomenon underlies a broad range of human pathologies, turning it into one of the most competitive and exciting research areas nowadays.
The number of diseases associated, directly or indirectly, with protein aggregation continues to grow. Perhaps the best known of these disorders are the neurodegenerative disorders, which include among others Alzheimer’s, Parkinson’s and Hungtinton’s diseases, the so-called prion diseases and amyotrophic lateral sclerosis (ALS). The abnormal aggregation of proteins is also behind pathologies like type II diabetes, cardiomyopathies, cataracts or even certain types of cancer [2, 3]. Some of these diseases have a clear genetic origin, but for most of them sporadic cases are the commonest. In some cases, transmission of the disease to healthy individuals has been reported, as in the case of prions. While aggregation-associated diseases can exhibit very different clinical manifestations, they share certain features; for example, they all display a late onset, with symptoms appearing usually in the adulthood, suggesting a common underlying mechanism of toxicity. Understanding the common and differential features behind these devastating diseases might allow progressing towards finding efficient therapeutic strategies.
Structural properties of protein aggregates
Proteins are the workhorses of the cell, being the final executors of the myriad of activities that sustained life requires. Generally, the polypeptide chains that come out from the ribosome in an unfolded conformation must adopt a defined three-dimensional structure -the native state– to be functional. In many cases, when this active conformation cannot be attained or maintained, misfolded proteins tend to self-assemble either intra- or extra-cellularly to build up insoluble deposits [4] (Figure 1). The formation of these aggregates promotes loss of protein function, saturates the protein quality control machinery, and leads to aberrant interactions and the subsequent co-aggregation of other essential cellular proteins. Thus, it is not surprising that protein aggregation becomes associated to pathological states. The proteins involved in these disorders do not share any sequential or structural similarity [3]. They can be intrinsically disordered, like α-synuclein in Parkinson, predominantly constituted by β-sheets such as SOD1 in ALS or, alternatively, α-helical like insulin in injection-localized amyloidosis. They can be small and monomeric, like amylin in type II diabetes or big and multimeric like transthyretin in familial polyneurophaty. Protein aggregation contradicts the one sequence / one structure / one function central biology dogma, because, independently of their sequence and native conformation, when these proteins aggregate into their toxic species they all converge to form macromolecular assemblies sharing a common fibrillar architecture, known as amyloid fibrils [5].
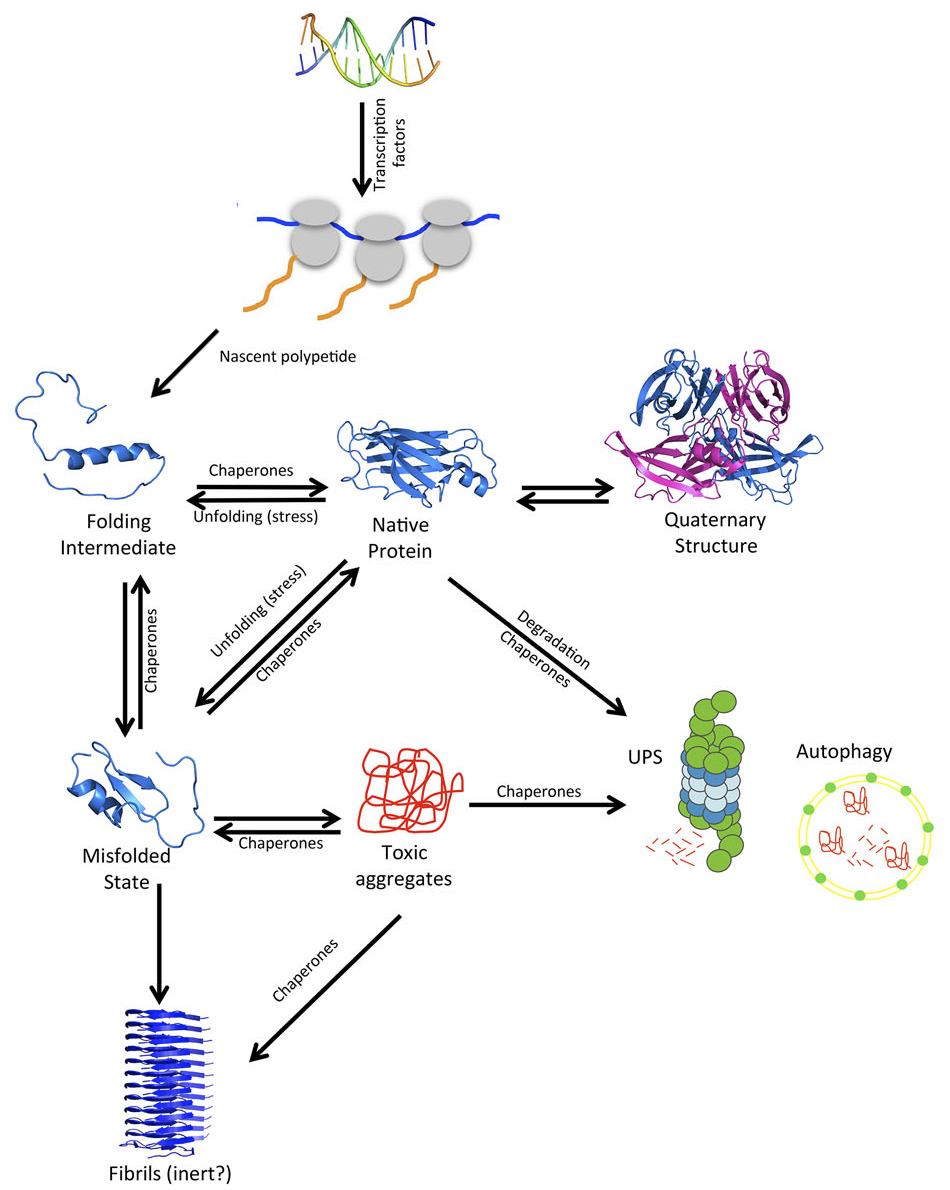
Figure 1. Overview of the proteostasis network (PN). A protein, during and after its synthesis at the ribosome, can adopt many different conformational states on the way to its native 3D structure. Imbalances in proteostasis often lead to protein aggregation and disease. Reproduced with permission from Pallarès & Ventura. Proteomics 2016, 16: 2570.
Amyloid fibrils are characterized by a polypeptide backbone organization in a cross-β disposition, which consists of a succession of contiguous β-strands stacked perpendicularly to the fibril axis. For many years, the characterization of amyloid fibrils using high resolution structural techniques like solution NMR and X-ray diffraction, remained elusive. Therefore, the presence of these insoluble deposits has been classically inferred from the results obtained using a battery of assays, including morphological analysis using transmission electron microscopy and atomic force microscopy, staining with amyloidtropic dyes such as Thioflavins and Congo Red, resistance to proteolysis, or checking out the seeding capacity characteristic of amyloid assemblies. Concomitantly, secondary structure analysis by circular dichroism, Fourier transformed infrared spectroscopy, or X-ray diffraction of aligned fibrils has been used to identify the characteristic cross-β-sheet signature in these protein aggregates [6]. Only recently, using short amyloid peptides able to form in the same conditions amyloid fibrils and microcrystals suitable for X-ray crystallography, the structure of the inner regions of amyloids could be studied at atomic resolution. In all cases, as predicted by low resolution techniques, the amyloid-like structures were shown to be formed by densely packed parallel or antiparallel intermolecular β-sheets [7]. However, the high resolution X-ray structure of a complete protein in its amyloid conformation remains to be elucidated. This structural gap has been partially filled by solid-state NMR studies, which exploiting extensive sets of experimental restraints have allowed to delineate the molecular interactions sustaining the amyloid fold in a reduced set of full-length proteins and peptides [8] (Figure 2).

Figure 2. Solid state NMR structure of HET-s in its prionic amyloid conformation. Each individual protein molecule contributes two layers of the β-sheet solenoid. The β-sheets are stacked orthogonal to the fibril axis.
Functional proteins cannot avoid aggregation
Contrary to what was initially thought, the phenomenon of amyloid formation is not restricted to a reduced number of proteins involved in disease. Instead, potentially any polypeptide is at risk of aggregation, and, indeed, it seems that ability to self-assemble into amyloid-like structures is an intrinsic property of polypeptide chains [9]. This surprise is not so striking now that we know that the the most stable conformation that a protein can adopt is not the native state, but the highly repetitive and densely stacked amyloid fibril. Hence, amyloid fibrils constitute a thermodynamic sink in which multiple proteins can get trapped [10]]. Indeed, computational studies at large scale indicate that the presence of short sequence stretches with high aggregation propensity is ubiquitous in all the analysed proteomes and some of them tend to be evolutionary conserved [11].
The questions that arise are: If protein aggregation impacts cellular fitness, why natural selection did not purge it out from the population, as it happens with deleterious mutations that impact the properties of structural or functional sites? Is there any benefit in conserving aggregation-prone regions in proteins that explains why is worth to take the risk? We are just beginning to find the answers to these questions by realizing that the establishment of protein functional interactions and the formation of anomalous contacts leading to the toxic protein aggregates are indeed two sides of the same coin, based on very similar physicochemical properties.
Protein regions with high aggregation propensity are usually rich in hydrophobic residues and depleted in charged amino acids. This composition is similar to the one of regions driving the formation of protein hydrophobic cores, which constitutes in many cases the first step of protein folding and whose correct assembly is crucial to maintain the metastable structure of functional proteins. Folding and aggregation kinetically compete in the cell, simply because aggregation-prone regions are essential to fold into functional protein structures in a biologically relevant time frame. This interrelationship is such that, in many cases, when we introduce amino acid changes intended to decrease the aggregation propensity of a given protein, we usually destabilize it or even prevent it to adopt a defined globular structure. We know now that there is a strong selective pressure to reduce the overall aggregation tendency of proteins and that nature has evolved a number of complementary structure- and sequence-based strategies to reduce the aggregation risk [12]. The fact that we still find such potentially dangerous regions in the cores of a large majority of proteins is a clear indication that functional globular proteins cannot avoid carrying a certain aggregation load during their lifetimes.

Figure 3. Therapeutics against Transthyretin (TTR) caused amyloidosis. Stabilization of the quaternary structure of a TTR mutant causing cardiomyopathy with chemical chaperones such as Tolcapone prevents protein dissociation, precluding aggregation and eliminating TTR toxicity in cardiomyocytes. Reproduced with permission from Sant’Anna, et al. Nat Commun. 2016, 7: 10787.
Aggregation-prone regions also play a crucial role in the formation of the quaternary structure of proteins or in the assembly of protein complexes [13]. This can be concluded from the fact that the interfaces between protein subunits display a higher aggregation propensity than exposed surfaces, which have evolved to minimize the aggregation risk by increasing the proportion of polar residues against the hydrophobic ones. Indeed, the presence of exposed hydrophobic residues on a protein surface is usually indicative that they play a functional role, as they have been shown to be more conserved than the rest of the amino acids [14]. Having exposed hydrophobic residues implies an inherent aggregation risk and, unless they serve for a specific purpose, they should have been purged during evolution. The unwanted dissociation of protein quaternary structures into their subunits is associated with the onset of a number of degenerative disorders, like the transthyretin (TTR) caused amyloidosis or ALS, just because the dissociated monomers freely expose aggregation-prone regions previously hidden at the interface, resulting in their fast self-assembly into amyloid like structures. The link between protein dissociation and aggregation provides a therapeutic window to halt the progression of these diseases. The stabilization of the quaternary structure of these proteins using small chemical compounds, known as chemical chaperones, is the only therapy we have nowadays in the market to target a protein aggregation-linked disease [15, 16] (Figure 3).
The prion-like phenomenon: generating revolutionary protein functions
When amyloid fibrils grow and divide with high efficiency they can propagate and are then termed prions. These fibrils propagate their conformation in a self-templating process. Prion diseases were thought to be exceptional because the pathology can be transmitted from organism to organism through a protein-based mechanism [17]. However, it is becoming apparent that protein-based propagation may reach beyond the scope of these relatively rare diseases to frequently occurring neurodegenerative disorders, including Alzheimer’s and Parkinson’s diseases [18]. These findings suggest a unifying mechanism underlying the pathogenesis of neurodegenerative disorders in which protein aggregates can be directly transmitted from pathologically affected to healthy, unaffected cells, thereby potentially extending the disease process throughout the nervous system [19].

Figure 4. Functional prion-like polymerization in human immune response. The mitochondrial protein MAVS, located on the surface of mitochondrial membranes, polymerizes into functional prion-like aggregates in response to viral infection. These assemblies activate and propagate an innate immune response in a reaction that depends on the recognition of viral RNA by the RIG-1 receptor. Reproduced with permission from Xu, H., et al. eLife 2014, 3: e01489.
Nevertheless, the traditional association of human prion proteins with disease has overshadowed one of the most interesting and unique attributes of prions: their ability to spontaneously shift between soluble and self-templating aggregated states. It is now clear that this property is exploited for functional purposes by different organisms and underlies some of the most revolutionary new concepts in biology, including protein- based genetic elements, membrane-free compartmentation, evolutionary capacitance and the revelation of cryptic genetic variation [20]. In these processes, typically, the prionic conformation compromises protein functionally, resulting in the expression of new phenotypes, previously repressed by the presence of the functional soluble conformation. However, for some prion-like proteins, a gain of function occurs and aggregation is used to propagate a biological function. In this way, the prionic conformation of the RNA-binding protein, Cytoplasmic Polyadenylation Element Binding protein (CPEB), displays increased affinity for RNA, contributing to long-term memory formation in metazoans [21]. Similarly, it has been shown that after viral infection, the human mitochondrial protein MAVS forms functional prion-like aggregates responsible for activating and propagating the innate immune response [22] (Figure 4). Importantly, protein response to environment based on protein conformational changes is much faster than receptor mediated activation of gene expression. Indeed, computational analyses indicate that prion-like proteins are present in the proteomes of organisms in all kingdoms of life [23], constituting a diverse and amazing group of proteins whose functional relevance would clearly expand beyond their potential link to pathology.
Conclusion
Protein aggregation is sustained because it is necessary to stablish and maintain both functional intra- and inter-molecular interactions. In addition, the formation of highly ordered macromolecular structures and the ability to shift between monomeric and assembled states allows to gain access to functions that are inaccessible to individual proteins. The potential formation of toxic aggregates and the subsequent development of late onset diseases seems to be the price we have to pay for that. The amazing new functions that we are uncovering for aggregated folds suggest that this price is not as high as we have always thought.
Institut de Biotecnologia i de Biomedicina – IBB,
Universitat Autònoma de Barcelona – UAB, Barcelona (Spain).
References
- Ventura S, Villaverde A. “Protein quality in bacterial inclusion bodies”. Trends Biotechnol, 2006, 24: 179. DOI: 10.1016/j.tibtech.2006.02.007.
- Invernizzi G, Papaleo E, Sabate R, Ventura S. “Protein aggregation: mechanisms and functional consequences”. Int J Biochem Cell Biol, 2012, 44: 1541. DOI: 10.1016/j.biocel.2012.05.023.
- Chiti F, Dobson CM. “Protein misfolding, functional amyloid, and human disease”. Annu Rev Biochem, 2006, 75: 333. DOI: 10.1146/annurev.biochem.75.101304.123901.
- Pallarès I, Ventura S. “Understanding and predicting protein misfolding and aggregation: Insights from proteomics”. Proteomics, 2016, 16: 2570. DOI: 10.1002/pmic.201500529.
- Fernàndez-Busquets X, de Groot NS, Fernandez D, Ventura S. “Recent structural and computational insights into conformational diseases”. Curr Med Chem, 2008, 15: 1336. DOI: 10.2174/092986708784534938.
- Sabate R, Ventura S. “Cross-β-sheet supersecondary structure in amyloid folds: techniques for detection and characterization”. Methods Mol Biol, 2013, 932: 237. DOI: 10.1007/978-1-62703-065-6_15.
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, Madsen AØ, Riekel C, Eisenberg D. “Atomic structures of amyloid cross-beta spines reveal varied steric zippers”. Nature, 2007, 447: 453. DOI: 10.1038/nature05695.
- Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. “Amyloid fibrils of the HET-s(218-289) prion form a beta solenoid with a triangular hydrophobic core”. Science, 2008, 319: 1523. DOI: 10.1126/science.1151839.
- Knowles TP, Vendruscolo M, Dobson CM. “The amyloid state and its association with protein misfolding diseases”. Nat Rev Mol Cell Biol, 2014, 15: 384. DOI: 10.1038/nrm3810.
- Jahn TR, Radford SE. “Folding versus aggregation: polypeptide conformations on competing pathways”. Arch Biochem Biophys, 2008, 469: 100. DOI: 10.1016/j.abb.2007.05.015.
- Linding R, Schymkowitz J, Rousseau F, Diella F, Serrano L. “A comparative study of the relationship between protein structure and beta-aggregation in globular and intrinsically disordered proteins”. J Mol Biol, 2004, 342: 345. DOI: 10.1016/j.jmb.2004.06.088.
- Monsellier E, Chiti F. “Prevention of amyloid-like aggregation as a driving force of protein evolution”. EMBO Rep, 2007, 8: 737. DOI: 10.1038/sj.embor.7401034.
- Castillo V, Ventura S. “Amyloidogenic regions and interaction surfaces overlap in globular proteins related to conformational diseases”. PLoS Comput Biol, 2009, 5: e1000476. DOI: 10.1371/journal.pcbi.1000476.
- Levy ED, De S, Teichmann SA. “Cellular crowding imposes global constraints on the chemistry and evolution of proteomes”. Proc Natl Acad Sci USA, 2012, 109: 20461. DOI: 10.1073/pnas.1209312109.
- Baranczak A, Kelly JW. “A current pharmacologic agent versus the promise of next generation therapeutics to ameliorate protein misfolding and/or aggregation diseases”. Curr Opin Chem Biol, 2016, 32: 10. DOI: 10.1016/j.cbpa.2016.01.009.
- Sant’Anna R, Gallego P, Robinson LZ, Pereira-Henriques A, Ferreira N, Pinheiro F, Esperante S, Pallares I, Huertas O, Almeida MR, Reixach N, Insa R, Velazquez-Campoy A, Reverter D, Reig N, Ventura S. “Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity”. Nat Commun, 2016, 7: 10787. DOI: 10.1038/ncomms10787.
- Prusiner SB. “Novel proteinaceous infectious particles cause scrapie”. Science, 1982,216: 136. DOI: 10.1126/science.6801762.
- Aguzzi A, Lakkaraju AK. “Cell Biology of Prions and Prionoids: A Status Report”. Trends Cell Biol, 2016, 26: 40. DOI: 10.1016/j.tcb.2015.08.007.
- Stopschinski BE, Diamond MI. “The prion model for progression and diversity of neurodegenerative diseases”. Lancet Neurol, 2017, 16: 323. DOI: 10.1016/S1474-4422(17)30037-6.
- Newby GA, Lindquist S. “Blessings in disguise: biological benefits of prion-like mechanisms”. Trends Cell Biol, 2013, 23: 251. DOI: 10.1016/j.tcb.2013.01.007.
- Si K, Choi YB, White-Grindley E, Majumdar A, Kandel ER. “Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation”. Cell, 2010, 140: 421. DOI: 10.1016/j.cell.2010.01.008.
- Xu H, He X, Zheng H, Huang LJ, Hou F, Yu Z, de la Cruz MJ, Borkowski B, Zhang X, Chen ZJ, Jiang QX. “Structural basis for the prion-like MAVS filaments in antiviral innate immunity”. eLife, 2014, 3: e01489. DOI: 10.7554/eLife.01489.
- Espinosa Angarica V, Ventura S, Sancho J. “Discovering putative prion sequences in complete proteomes using probabilistic representations of Q/N-rich domains”. BMC Genomics, 2013, 14: 316. DOI: 10.1186/1471-2164-14-316.