
Super-resolution optical nanoscopy gets the 2014 Nobel Prize in Chemistry
 Optical microscopy is without any doubts the most important imaging tool for biologists. Unfortunately, Ernst Abbe‘s nineteenth century formulation of the resolution limit in microscopy let generations of scientists believe that optical resolution at the nanometer scale was impossible. Remarkably, within 2004 and 2006 several optical techniques were invented and the diffraction limitation was overcome! In October 2014, the Nobel Prize in Chemistry was awarded to W. E. Moerner, E. Betzig, and S. W. Hell for the development of super-resolution fluorescence microscopy. The jury members stated that super-resolution nanoscopy…
Optical microscopy is without any doubts the most important imaging tool for biologists. Unfortunately, Ernst Abbe‘s nineteenth century formulation of the resolution limit in microscopy let generations of scientists believe that optical resolution at the nanometer scale was impossible. Remarkably, within 2004 and 2006 several optical techniques were invented and the diffraction limitation was overcome! In October 2014, the Nobel Prize in Chemistry was awarded to W. E. Moerner, E. Betzig, and S. W. Hell for the development of super-resolution fluorescence microscopy. The jury members stated that super-resolution nanoscopy…
…is expected to revolutionize biology and medicine allowing for realistic, quantitative descriptions at nanoscale resolution of the dynamics of the complex, multidimensional molecular biological processes that define the phenotypes of all life forms.
In here we briefly summarize how these forms of super-resolution nanoscopy work, starting with a short description of the diffraction limit of light.
The diffraction limit of light
Towards the end of the nineteenth century Ernst Abbe and Lord Rayleigh formulated what is commonly known as the diffraction limit for microscopy. Roughly speaking, this limit states that it is impossible to resolve two elements of a structure which are closer to each other than about half the wavelength (λ) of the light used for observation in the lateral (x,y) plane, and even further apart in the normal (z) direction. The immediate consequence of the diffraction limitation is that it is not possible to focus a laser beam to a spot of dimensions smaller than about λ/2. At visible wavelengths (λ= 470-670 nm) the maximum optical resolution one can obtain is thus between 250-350 nm. However, as many structures in Nature, including biological objects, have dimensions much smaller than these values, it has been impossible until recently to visualize them in the natural context of living cells. The major breakthrough of super-resolution techniques is that they allow the capture of images with an apparent resolution higher than the diffraction limit. Curiously enough, the techniques that warranted the Nobel Prize in Chemistry do not really break the diffraction limit of light!
Breaking the diffraction limit
Super-resolution techniques can be classified into two broad categories, true super-resolution techniques, which capture information contained in evanescent waves, i.e., in the near-field regime, and functional super-resolution techniques, which work in the conventional far-field regime and use clever experimental techniques and known properties of fluorescent emitters to reconstruct a super-resolution image.
Curiously enough, the techniques that warranted the Nobel Prize in Chemistry do not really break the diffraction limit of light!
Within the true super-resolution techniques, the most conceptual way to break the diffraction barrier is using a light source and/or a detector with nanometer dimensions. In fact, diffraction is truly a far-field effect: The light from an aperture is the Fourier transform of the aperture in the far-field. In the near-field, this is not necessarily the case. Near-field scanning optical microscopy (NSOM) is a technique that funnels light through a tiny tip of a pulled fiber — and the aperture can be on the order of tens of nanometers. When the tip is brought to nanometers away from an object, the resolution is limited not by diffraction but by the size of the tip aperture, and an image can be built by raster scan of the tip over the object surface.
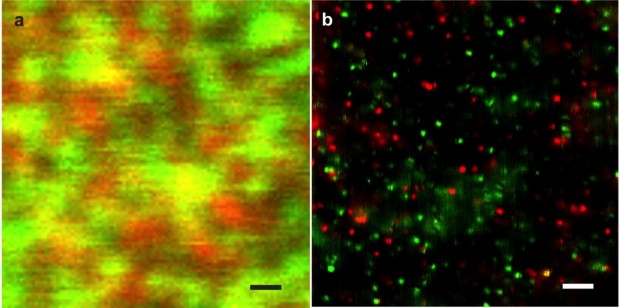
Figure 1. a) Confocal image of two different types of fluorescent molecules (red & green emission). The diffraction limit of confocal does not allow discrimination of individual molecules so that the image appears blurred. b) Dual color super-resolution NSOM image of the same sample using a photonic antenna. Each colored spot corresponds to an individual molecule. Optical resolution is 20 nm. Scale bars are 1um. Published in: Mivelle M, van Zanten TS, Garcia-Parajo MF. “Hybrid photonic antennas for subnanometer multicolor localization and nanoimaging of single molecules”. Nano Lett, 2014, 14: 4895.
Unfortunately NSOM suffers from the low light throughput of the illumination source. Moreover, it is technically challenging to implement, so that its applicability to the biological domain has remained quite limited. On the positive side of the balance and as a truly super-resolution technique, NSOM does not rely on the fluorescent properties of emitters, providing quantitative information at the nanometer scale. Subsequent advancements from the nanophotonics field, and in particular the use of photonic antennas, can overcome the main limitation of light throughput associated with NSOM. Indeed, real 20 nm optical resolution and localization accuracies at the sub-nanometer level have been recently demonstrated by engineering optical antennas at the apex of optical fibers (see Figure 1).
Super-resolution in the far-field regime
Functional super-resolution optical techniques work in the far-field regime and as such are per-se diffraction limited. How can one thus obtain a super-resolution image if diffraction still dominates? The trick is to combine standard far-field optics with careful manipulation of the photo-physical properties of the fluorescence markers used to label the sample. No wonder then, why the Nobel Prize was given in Chemistry and not in Physics.
The trick is to combine standard far-field optics with careful manipulation of the photo-physical properties of the fluorescence markers used to label the sample.
In general terms, functional super-resolution techniques rely on the controlled switching of the sample fluorescence emission in time, either spatially, as conceived in stimulation emission depletion (STED) nanoscopy, or stochastically as implemented in single molecule localization methods. Stefan Hell (one of the Nobel winners) and colleagues conceptually introduced STED more than a decade ago. The technique is based on creating a nanometric optical region by first exciting fluorophores to an excited state over a diffraction-limited region using a pulsed laser. A second pulsed laser illuminates the sample with a doughnut-shape like pattern, in a wavelength that depletes the excited state of the fluorescent molecules back to the ground state. Fluorescence is effectively detected only from the hole of the doughnut, and the final spot size can be tuned to balance resolution against signal. The overlapped excitation and depletion beams are then raster scanned over the sample, delivering images with resolutions typically between 30 to 80 nm (see Figure 2).

Figure 2. Super-resolution STED image of the pathogen recognition receptor on the cell membrane of an immature dendritic cell. Resolution is 95 nm. Published in: Torreno-Pina JA, Castro B, Manzo C, Buschow S, Cambi A, Garcia-Parajo MF. “Enhanced receptor-clathrin interactions induced by N-glycan mediated membrane micropatterning”. Proc Nat Acad Sci USA, 2014, 111: 11037.
Single molecule localization methods on the other hand, rely on the stochastic on-off switching of individual molecules. The essence of the method is allowing only a subset of fluorescent molecules to be photoactive at a given time and ensuring that the nearest-neighbor distance between active molecules is larger than the diffraction limit. Methods that make use of this principle are photoactivatable localization microscopy (PALM/FPALM) and stochastic optical reconstruction microscopy (STORM). The basic premise of both techniques is to fill the imaging area with many dark fluorophores that can be photoactivated into a fluorescing state by a flash of light. Because photoactivation is stochastic, only a few, well separated molecules “turn on.” Then Gaussians are fit to their point spread functions (PSF) to high precision. After the few bright dots photobleach, another flash of the photoactivating light activates random fluorophores again and the PSFs are fit of these different well-spaced objects. This process is repeated many times, building up an image molecule-by-molecule. Because the molecules were localized at different times, the “resolution” of the final image can be much higher than that limited by diffraction. The main difference between PALM and STORM resides on the type of fluorophores used for photoactivation: PALM relies on autofluorescent proteins, while STORM uses organic switchable dyes. The ascertainable localization accuracy depends strongly on the total number of photons being detected and can be as high as 10 nm (Figure 3).

Figure 3. Comparison of wide-field (upper) and STORM (lower) images of the core histone protein H2B in an intact human fibroblast nucleus. Image courtesy of Ricci MA, Manzo C, Garcia-Parajo MF, Lakadamyali M, Cosma MP, at ICFO, Barcelona (Spain).
Two major ingredients have been key to the development of single molecule localization methods. First, the capability to detect individual molecules; Second, their control in terms of on-off switching. Both E. Betzig and W. E. Moerner (Nobel awardees) set the floor for the development of these techniques. Already in 1993 Betzig demonstrated that individual fluorescent molecules could be detected at room temperature using NSOM. One year later, the detection of individual molecules was also demonstrated using less complicated optical techniques, such as confocal or wide field microscopy. However, molecules could only be detected at the single level provided that they were physically separated by distances larger than the diffraction limit of the microscope.
Moerner made a major discovery in 1997: The emission of photons by fluorescent molecules is interleaved with dark periods
W.E. Moerner who had already demonstrated the detection of individual molecules at cryogenic temperatures, made a major discovery in 1997: Fluorescent molecules, and in particular, autofluorescent proteins do not emit their photons in a stable fashion. Instead their emission is interleaved with dark periods where no fluorescence is detected. This on-off behavior is known as blinking. The next challenge that led to the final development of super-resolution based on localization of individual molecules was the control of these on-off switching events. In 2006 two separate papers appeared almost simultaneously: both E. Betzig and S. Hess succeeded on controlling the on-off switching of autofluorescent properties and invented PALM. One year later, the group of X. Zhuang managed to control the on-off switching of fluorescent dyes, leading to the invention of STORM. One might really wonder why S. Hess and X. Zhuang did not share the Nobel Prize together with Hell, Betzig and Moerner …
A short history with a promising future
As stated by Prof. Ehrenberg, member of the Nobel Committee for Chemistry,
The history of super-resolved fluorescence microscopy is short. The ensemble-fluorophore STED-microscopy was implemented in the year 2000 and single-fluorophore based methods in the year 2006. In spite of this, the rapidly developing techniques of super-resolved fluorescence microscopy are already applied on a large scale in major fields of the biological sciences, like cell biology, microbiology and neurobiology. At this point there is all reason to forecast that this development, already producing hosts of novel and previously unreachable results, will accelerate over the next decades.
Institut de Ciencies Fotoniques – ICFO
Mediterranean Technology Park
08860 Castelldefels (Barcelona), Spain.
Institució Catalana de Recerca i Estudis Avançats – ICREA
08010 Barcelona, Spain.
E-mail: maria.garcia-parajo@icfo.eu
References
See: Official Web site for the Nobel Prize: http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2014/
Key papers on super-resolution nanoscopy
Dickson RM, Cubitt AB, Tsien RY and Moerner WE. “On/off blinking and switching behaviour of single molecules of green fluorescent protein”. Nature, 1997, 388: 355.
Klar TA, Jakobs S, Dyba M, Egner A, Hell SW. “Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission”. Proc Natl Acad Sci USA, 2000, 97: 8206.
Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. “Imaging intracellular fluorescent proteins at nanometer resolution. Science, 2006, 313:1642.
Hess ST, Girirajan TP and Mason MD.”Ultra-high resolution imaging by fluorescence photoactivation localization microscopy”. Biophys J, 2006, 91:4258-4272.
Rust MJ, Bates M and Zhuang X. “Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM)”. Nat Methods, 2006, 3:793.
Huang B, Babcock H and Zhuang X. “Breaking the diffraction barrier: Super-resolution imaging of cells”. Cell, 2010, 143: 1047.
Hinterdorfer P, Garcia-Parajo MF, Dufrêne YF. “Single-molecule imaging of cell surfaces using near-field nanoscopy”. Acc Chem Res, 2012, 45: 327.